器审中心网站法规文件专栏列明了医疗器械注册相关所有法规、部门规章、规范性文件。申请人可查询下载自学《医疗器械监督管理条例》《医疗器械注册与备案管理办法》《体外诊断试剂注册与备案管理办法》等一系列法规、部门规章以及其他规范性文件。
器审中心网站开设“器审云课堂”、“CMDE Learning Online”中英文课程栏目,相关课程亦可在“中国器审”微信公众号和学习强国app进行学习。
注册申报流程有哪些环节?从哪里了解每个环节的要求?
器审中心网站“办事大厅”下“办事指南”栏目列举了一类备案、注册申报和技术审评流程简图中英文版。
其中,注册申报流程可以查看“注册申报流程简图”。简图中列出了注册申报过程中可能涉及到的环节和事项,可以点击查看各个环节点,了解相关的概念、工作程序、一些实用小提示,可通过点击链接下载。如果是进口一类产品,则可以点击同在“办事大厅”栏目下的“一类备案流程简图”了解相关内容。对于手机端,在“中国器审”公众号“企业服务”栏下点击“申报流程”即可查看。
“中国器审”公众号“器审要闻”栏下“注册图说”栏目,还可查看包含注册申报流程的各流程图解以及某些具体事项的图解。
在具体产品的研发与注册时是否有全面的技术指导文件可查询?遇到技术和法规问题,有哪些途径可以寻求答案?
(一)指导原则、审评要点与标准及临床评价路径
器审中心网站“审评科学”专栏可查询所有中国指导原则与FDA及欧盟相关指导原则文本。在“指导原则”下“分类目录-指导原则-标准”栏目点开具体产品,该产品相关的专用指导原则、审评要点、适用标准、临床评价路径均一一列明,并提供指导原则、审评要点、标准等技术文件的下载,供申请人参考。
以微波手术设备为例,网站提供的信息如下图,对于研发企业可通过这种方式直接获取与此类产品相关的所有指导技术文件:

(二)常见共性问题解答
针对审评中的常见问题,器审中心网站审评科学专栏下“共性问题”专栏及“中国器审”公众号“信息公开”栏目“答疑解惑”专栏均日常更新。既包括法规指导原则执行层面的解读,也包括具体产品技术问题。申请人遇到问题时可先行查询。
例如,对有源器械的使用期限研究资料有疑问,可在“中国器审”公众号“答疑解惑”栏目中查询到以下内容:
(三)监管科学相关研究进展及思考
器审中心网站“审评科学”专栏下“审评论坛”专栏及“中国器审”公众号“信息公开”栏目“审评论坛”专栏均日常更新审评相关研究进展及思考,对于相关产品研发可查询参考。如《血管介入器械表面润滑涂层风险评估及监管探索 》《骨填充材料的分类及发展现状概述》《无源非植入医疗器械包装注册监管的思考》《当前关于病原体宏基因组高通量测序产品的几点考虑》等。
(四)审评报告公开
创新、优先、同品种首个产品均公开审评报告,可在器审中心网站和公众号“信息公开-审评报告”栏目下查询参考。
(五)咨询
如网站和公众号上述内容均无法解答相关疑问,可通过咨询解决。具体咨询路径见下文关于咨询路径的相关内容。
体外诊断产品临床前申报资料可以参考的指导原则有哪些?
通用指导原则包括《定性检测体外诊断试剂分析性能评估注册审查指导原则》《定量检测体外诊断试剂分析性能评估注册审查指导原则》《体外诊断试剂参考区间确定注册审查指导原则》《质控品注册审查指导原则——质控品赋值研究》《体外诊断试剂说明书编写指导原则》《医疗器械产品技术要求编写指导原则》等。
另外,还可参考具体产品的指导原则。
在器审中心网站 “审评科学”栏目下点击“临床评价路径推荐”,三个通告内容包含了医疗器械分类目录中产品的临床评价推荐路径,分别为:关于发布《医疗器械分类目录》子目录01、04、07、08、09、10、19、21相关产品临床评价推荐路径的通告(2022年第30号)、关于发布《医疗器械分类目录》子目录02、03、05、06、16、18、20相关产品临床评价推荐路径的通告(2022年第24号)、关于发布《医疗器械分类目录》子目录11、12、13、14、15、17、22相关产品临床评价推荐路径的通告(2022年第20号)。申请人可参考上述文件确定产品的临床评价路径。产品是否开展临床试验,应参照《决策是否开展医疗器械临床试验技术指导原则》进行判定。
体外诊断试剂产品是否需开展临床试验按照《免于进行临床试验体外诊断试剂目录》执行。列入目录的产品按照同品种比对方式开展临床评价,未列入目录的二、三类体外诊断试剂需开展临床试验。
(一)医疗器械产品:
开展临床试验应参照法规《医疗器械临床试验质量管理规范》、通用指导原则包括《医疗器械临床评价技术指导原则》《医疗器械临床评价报告撰写技术指导原则》《医疗器械临床试验设计指导原则》《接受医疗器械境外临床试验数据技术指导原则》《医疗器械临床试验数据递交要求注册审查指导原则》以及具体产品指导原则。
(二)体外诊断产品:
开展临床试验应参照法规《医疗器械临床试验质量管理规范》、通用指导原则包括《体外诊断试剂临床试验技术指导原则》《接受医疗器械境外临床试验数据技术指导原则》《使用体外诊断试剂境外临床试验数据的注册审查指导原则》《体外诊断试剂临床试验数据递交要求注册审查指导原则》《来源于人的生物样本库样本用于体外诊断试剂临床试验的指导原则》以及具体产品的指导原则。
在器审中心网站“审评科学”栏目下点击“指导原则”,进入“分类目录--指导原则-标准”-23通用”,参照《医疗器械临床评价等同性论证技术指导原则》、《决策是否开展医疗器械临床试验技术指导原则》、《医疗器械临床试验设计指导原则》、《接受医疗器械境外临床试验数据技术指导原则》、《医疗器械注册申报临床评价报告技术指导原则》、《医疗器械临床评价技术指导原则》等指导原则撰写临床评价报告。
对免于临床试验的体外诊断试剂,进行临床评价可参考《免于临床试验的体外诊断试剂临床评价技术指导原则》。
在“中国器审”公众号“企业服务”栏下点击“进度查询”或在器审中心网站“办事大厅”栏下点击“审评进度查询”输入受理号等信息即可查询本单位申报产品的审评进度。
在“中国器审”公众号“企业服务”栏下点击“产品跟踪”按要求绑定企业信息和产品后,产品审评环节变化时可以通过微信收到主动推送的状态变化通知。
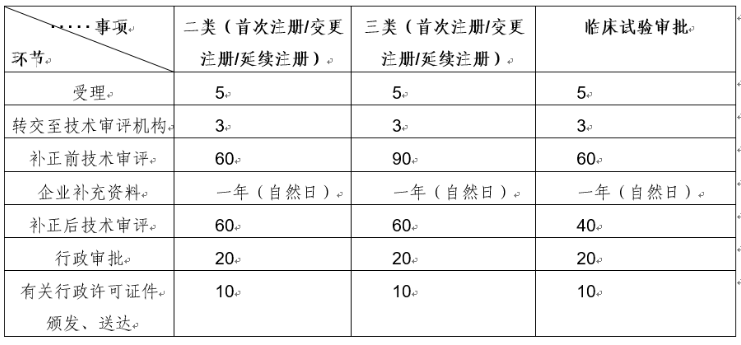
根据《医疗器械注册与备案管理办法》(国家市场监督管理总局令第47号)和《体外诊断试剂注册与备案管理办法》(国家市场监督管理总局令第48号)中有关工作时限要求,相关各环节法定时限详见下表:
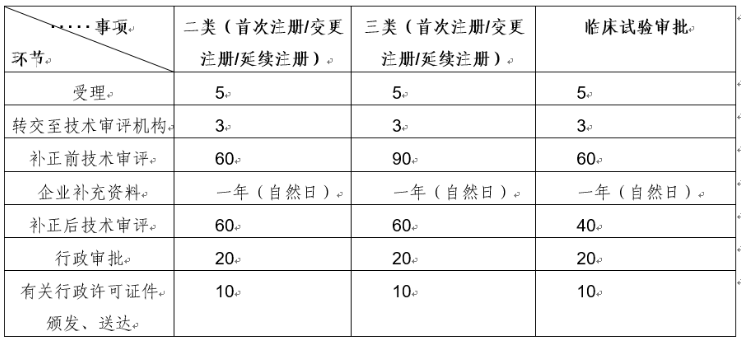
注:上表未标明自然日的时限均为工作日。
器审中心持续深化审评制度改革,不断强化审评能力建设,努力缩短审评工作用时。根据中心统计数据,2022年器审中心的审评工作总体用时按管理类别看,二类产品注册审评总体平均用时为72个工作日,三类产品注册为99个工作日;按申请事项看,产品首次注册审评总体平均用时为95个工作日,变更注册为64个工作日,延续注册为39个工作日。以上用时为当前统计平均用时,供大家参考。
需注意的是,以下时间不计入上述相关工作时限:
(一)申请人补充资料、核查后整改等所占用的时间;
(二)因申请人原因延迟核查的时间;
(三)外聘专家咨询、召开专家咨询会、药械组合产品需要与药品审评机构联合审评的时间;
(四)根据规定中止审评审批程序的,中止审评审批程序期间所占用的时间;
(五)质量管理体系核查所占用的时间。
器审中心咨询途径有哪些?在研发申报的各个阶段如何咨询?
器审中心对行业提供全流程咨询服务,申请人可在不同的阶段选择适用的咨询方式解决问题。具体咨询方式可在“中国器审”公众号“企业服务”“申报流程”各环节点击查询。
具体各时间阶段对应的咨询路径如下:详情请点击此处。